
文獻標識碼: A
文章編號: 0258-7998(2013)09-0135-04
結構生物信息學已成為當前計算機科學領域的一個研究熱點。其主要研究目標是獲取生物系統的高分辨率結構信息,從而精確地推理系統的功能、預測某些修改或擾動對系統造成的影響。與生物信息學的其他領域相比,結構生物信息學面臨眾多新的挑戰。其中之一是多數結構生物信息學問題的搜索空間是連續的;換言之,生物系統的微觀結構一般通過原子在空間坐標系中的位置來表示,而坐標通常是連續變量,因此搜索空間是無窮的。雖然某些近似方法可以在一定程度上應對結構生物信息學問題的這種連續特性,但是從搜索空間中求得最終解仍然需要進行大量的運算[1]。
DNA-蛋白質匹配是生物信息學的一個重要問題。傳統的序列分析方法往往會導致較高的多重檢驗錯誤率(false-positive rate)[2]。多數基于結構生物信息學的DNA-蛋白質匹配方法把獲得轉錄因子-DNA組合體的信息作為一個必備條件,而轉錄因子-DNA組合體的信息通常需要通過實驗獲得,這種實驗通常需要耗費大量的時間。參考文獻[2]提出的基于退火算法的匹配算法避開了這個問題,但是使用該方法進行DNA-蛋白質匹配仍需要較大的運算量。參考文獻[3]基于參考集索引技術提出了一種快速的序列分析算法,并將其應用于DNA-蛋白質匹配。參考文獻[4]采用多CPU的方式設計實現了并行化的DNA-蛋白質序列分析方法,并取得了較高的加速比。但上述工作均未涉及基于結構生物信息學的匹配方法的加速問題。
應對大運算量問題的一個常用方法是并行計算。NVIDIA公司統一計算設備架構CUDA(Compute Unified Device Architecture)是一種全新的并行計算架構。在CUDA的支持下,通用計算圖形處理單元GPGPU(General Purpose Graphic Processing Unit)可以作為一種通用的效率、硬件加速器,發揮出強大的運算能力[5]。
本文針對參考文獻[2]的DNA-蛋白質匹配方法,從計算的角度對其進行分析,設計實現并優化了基于CUDA的加速方法,通過實驗驗證了其加速性能。
1 相關知識介紹
1.1 基于退火算法的DNA-蛋白質匹配算法
給定一條DNA鏈和一條蛋白質鏈,兩者之間的相對位置存在很多種可能。假定兩者均為剛體,DNA-蛋白質組合體的物理能量主要受兩者相對位置的影響,組合體的物理能量越低,其穩定性越強。該方法試圖尋找使得組合體最穩定的相對位置,這一結果對生物信息學的研究具有重要意義。
從計算的角度看,該方法的基礎是退火算法。為了提高搜索精度,該匹配方法通常需要隨機產生大量初始“種子”,為每個“種子”啟動一個基于退火算法的搜索過程,在整個解空間中尋找最穩定的相對位置。這種匹配方法的流程如圖1所示。

圖1中的初始值為T0,溫度T及溫度衰減周期interval為正整數,溫度衰減系數為a。每當算法所經歷的平移和旋轉次數達到interval的非零整數倍時,T衰減為原來的a倍(0<a<1),Tmin為溫度的最小值。
DNA-蛋白質組合體的物理能量E1、E2分別代表第n次(1≤n≤Smax,Smax為平移和旋轉次數的最大值)平移和旋轉前后組合體的物理能量。Emin代表通過算法得到的組合體物理能量的最小值。如果E2<E1,則接受第n次平移和旋轉的結果;否則,計算指數式exp(-(E2-E1)/T)的值,并使用C++庫函數drand48( )產生一個在區間[0,1]上均勻分布的隨機數。如果指數式的值小于隨機數的值,則接受第n次平移旋轉的結果,反之不接受。
step為當前進行到了第幾次平移和旋轉。
1.2 CUDA編程模型及線程調度方式的特點
CUDA程序通常包含CPU串行代碼和GPU并行代碼,后者被稱作CUDA程序的內核。在執行內核時,CUDA會產生大量并行工作的線程,以單指令多數據SIMD(Single Instruction Multiple Data)方式完成整個內核的計算任務。CUDA采用網格(grid)和線程塊(block)來組織線程[6]。一個block的線程又被劃分為一個或多個線程組(warp)。warp是CUDA程序最基本的調度單位,屬于同一個warp的各個線程會從CUDA程序代碼段中相同的程序地址同時開始執行,但是各線程具有獨立的當前指令指針和寄存器狀態。因此,各線程可能會有不同的執行路徑,例如執行不同的分支選擇結構代碼或循環結構代碼[7]。warp執行特點如圖2所示。

假設一個warp中共有4個線程:線程1~線程4,它們的執行時間分別是t1~t4,并且t3<t1<t2<t4。因為CUDA的基本調度單位是一個完整的warp而非單個線程,所以整個warp的執行時間取決于執行時間最長的線程t4。其他3個線程在執行完畢后必須等待還沒有執行完畢的線程,因此有一段時間處于空閑狀態,相應的計算資源也就被閑置了。例如,線程3閑置的計算資源最多,其空閑時長為(t4-t3)。造成計算資源閑置的原因是同一warp中各個線程的執行路徑不同;而CUDA采用的是SIMD并行方式,執行路徑的不同是由每個線程所處理的數據差異造成的。因此,依照一定規則對數據進行重新排列是減少這種計算資源閑置狀況的常用方法[8]。重排規則通常視具體應用而定。
2 使用CUDA加速DNA-蛋白質匹配
2.1 從計算角度分析匹配算法
匹配方法的流程已由圖1給出,除參數初始化外,該方法可分為三部分: (1)平移、旋轉DNA和蛋白質;(2)計算DNA-蛋白質組合體的能量;(3)后續處理。這3部分在普通CPU平臺上所耗時間依次為: 73.624 s、1 138.945 s、110.809 s(僅使用一個隨機初始種子,退火算法參數的預設值及軟硬件平臺參數指標見本文第3部分),實驗使用的DNA、蛋白質結構數據來自參考文獻[9]編號為2GLI的文件。其他的DNA、蛋白質結構數據的實驗結果也顯示了計算能量所耗費的時間遠超過其他兩個部分。計算能量時,DNA-蛋白質組合體所處的三維空間被劃分成若干個棱長為埃米(10-10 m)級的晶格。一條DNA鏈由若干個DNA原子構成,一條蛋白質鏈由若干個蛋白質原子構成;以2GLI組合體為例,共有855個DNA原子和1 270個蛋白質原子。每次平移和旋轉前后,每個原子所屬的晶格都可能發生改變,每個晶格所包含的原子個數也可能發生改變。Di(i=1,2,…,ND),Pj(j=1,2,…,NP)分別代表組合體中的DNA原子數量和蛋白質原子數量。DNA原子i(i=1,2,…,ND)在三維空間中所處的晶格和相鄰的晶格共有27個,統稱為原子i的臨近晶格(Neighboring Lattice),以pi,j(x,1,2,…,ni)代表這27個晶格中的所有蛋白質原子,這些原子即原子i的相鄰蛋白質原子。以di,j代表DNA原子i與蛋白質原子jx之間的歐式距離,兩個原子之間的能量是di,j的函數:E(di,j)。則組合體的總能量為:
通過累加各線程的能量部分和可以得到組合體的總能量。另外,如果B<ND,則需要把ND個DNA原子平均分成「ND/B?骎塊(最后一塊可能不足B個原子),然后,由整個block的線程依次處理各塊,算法2第3行的循環結構即為了達到這個目的。
2.3 優化并行算法
考慮到本文1.2節所述warp的執行特點,上述并行方式存在一定的不足。算法2中第7行的循環次數取決于當前晶格中蛋白質原子的個數,同一warp中各線程的循環次數可能會不同,如果差異過大,會造成嚴重的計算資源閑置;對所有線程而言,算法2第3行、第6行循環結構的循環次數是確定的。
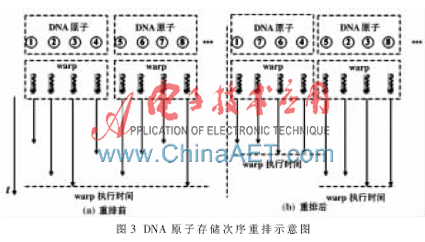
假設DNA存儲在一個數組中,且該數組位于GPU主存(global memory)中。為了盡量減少計算資源閑置,重排DNA原子的原有次序(DNA鏈中的次序),處于同一個晶格中的DNA原子被存儲在數組的某一段連續區域內。采用這種優化措施是基于以下原因:(1)由式(1)可知,總能量由E(di,j)做算術加法得到,與E(di,j)的計算次序無關;(2)為了提高GPU主存的帶寬利用率,同一warp中的線程通常從主存中的臨近區域讀取數據,即內存訪問對齊(coalescing)[6];(3)DNA的雙螺旋結構是非線性的,DNA在鏈中相鄰的原子未必處于同一晶格中;(4)同一晶格中的DNA原子的臨近蛋白質原子數量相同,重排可以減少因線程執行路徑差異造成的計算資源閑置。重排的效果如圖3所示。
3 實驗
3.1 實驗平臺
硬件平臺:Intel CoreTM2 E7500 CPU,2.93 GHz,NVIDIA GTX 660圖形處理器(單個warp包含 32個線程),CPU主存4 GB。
軟件平臺:Linux操作系統內核版本2.6.18-194.el5,gcc版本4.1.2,CUDA版本5.0。
3.2 實驗參數設置與結果
DNA-蛋白質結構數據編號為2GLI。CPU版程序以單線程串行方式執行;GPU版程序block size固定為512;各block從不同的初始種子出發,分別基于退火算法進行搜索,每個block對應一個初始種子。退火算法參數:初始溫度T0=120℃,最低溫度Tmin=0.001℃,溫度衰減周期interval=800,溫度衰減系數a=0.99,最大平移旋轉次數Smax=106。
實驗結果如表1所示。與單線程CPU程序相比,未經優化的GPU程序將獲得最高可達8倍左右的加速比;而經過重排優化后,加速比在此基礎上進一步顯著提升,最高可達15倍左右。
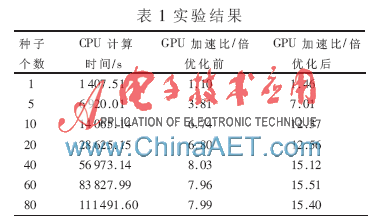
本文針對一種典型的DNA-蛋白質匹配算法,設計實現了基于CUDA的并行化方法,從線程調度的角度對該方法進行優化,并通過實驗驗證了加速性能。實際應用往往需要為一個組合體產生大量的初始種子(數百個),并為每個種子開啟一個基于退火算法的搜索過程;其目的是達到較高的匹配精度。實驗顯示,使用單個GPGPU加速,在單個block包含的線程數不變的前提下,隨著初始種子數量的增加,加速比逐漸趨于穩定。例如,當初始種子個數超過40后,加速比基本穩定在15倍左右。其原因在于單個GPGPU的計算能力存在上限,當種子足夠多時,其計算能力已得到較充分利用,無法繼續提高加速比。為了滿足實際應用的需求,下一步的工作將考慮使用基于GPGPU的集群來加速匹配算法,以進一步提高加速比。
參考文獻
[1] BOURNE P E,WEISSIG H. Structural bioinformatics[M]. Hoboken: Wiley-Liss Inc, 2003.
[2] Liu Zhijie, Guo Juntao, Li Ting, et al. Structure-based prediction of transcription factor binding sites using a protein-DNA docking approach[J]. Proteins, 2008,72(4):1114-1124.
[3] 戴東波,熊赟,朱揚勇.基于參考集索引的高效序列相似性查找算法[J].軟件學報,2011(4):718-731.
[4] Zhang Zhang, Xiao Jingfa, Wu Jiayan, et al. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments[J]. Biochemical Biophysical Research Communications, 2012,419(4):779-781.
[5] 徐新海,楊學軍,林宇斐,等.一種面向CPU-GPU異構系統的容錯方法[J].軟件學報,2011,22(10):2538-2552.
[6] NVIDIA Cooperation.CUDA programming guide version 5.0[EB/OL].[2013-05-15].http://docs.nvidia.com/cuda/cudac-programming-guide/.
[7] TIANYI D H,TAREK S A. Reducing branch divergence in GPU programs[A]. In Proceedings of the Fourth Workshop on General Purpose Processing on Graphics Processing Units[C]. London: ACM.2012.
[8] IMEN C, AHCENE B, NOUREDINE M. Reducing thread divergence in GPU-based b&b applied to the flow-shop problem[A]. In Proceedings of the 9th International Conference on Parallel[C]. Berlin: Springer-Verlag.2011.
[9] Rutgers and UCSD. Protein Data Bank [DB/OL].[2009-02-24].http://www.rcsb.org/pdb/explore/explore.do?structureId=2gli.
[10] GENE A. Validity of the single processor approach to achieving large-scale computing capabilities[A]. In Proceedings of the April 18-20(AFIPS′67)[C]. New York:ACM.1967.